2.1 UDMH分子C70上的吸附行为
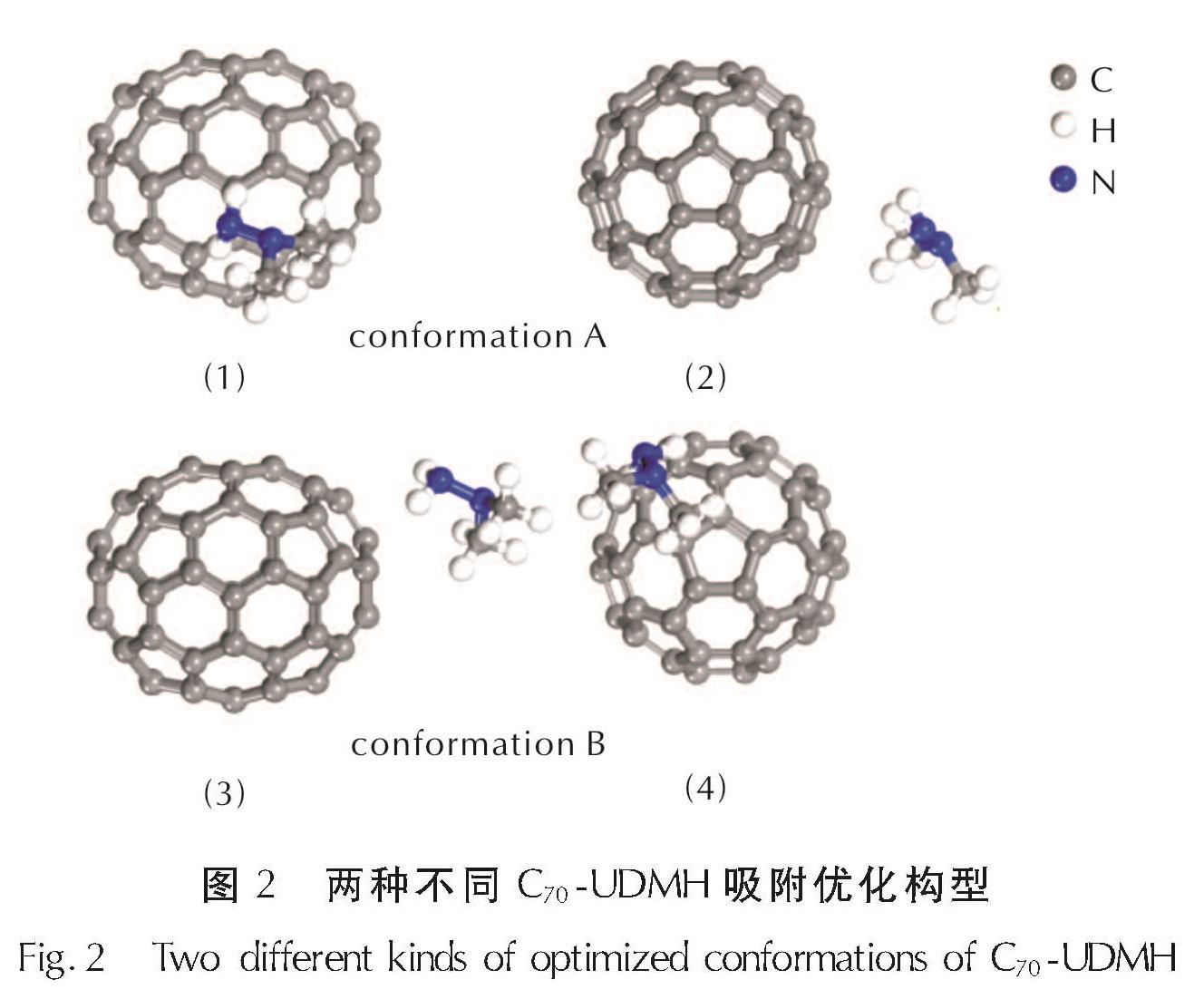
C70团簇的分子结构存在一定的对称性[18,19],故仅考虑少数位点作为吸附位点。经结构优化得到两种不同的吸附构型如图2所示。规定图1(a)为C70正面图像,图2中(1)、(2)分别为A构型的正视图与侧视图;(3)、(4)为B构型的正视图与侧视图。
如图2所示,相对于C70分子,A构型中UDMH分子的NH2基团和CH3基团均位于C70六元环的上方; B构型中NH2基团位于C70分子中六元环上方,CH3基团则位于五元环上方。此外,A构型中UDMH分子中NH2基团上N原子与C70分子的最短距离为3.396Å,B构型中的最短距离为3.645Å,较长的距离意味着两个分子之间的氢键较弱。
图2 两种不同C70-UDMH吸附优化构型
Fig.2 Two different kinds of optimized conformations of C70-UDMH
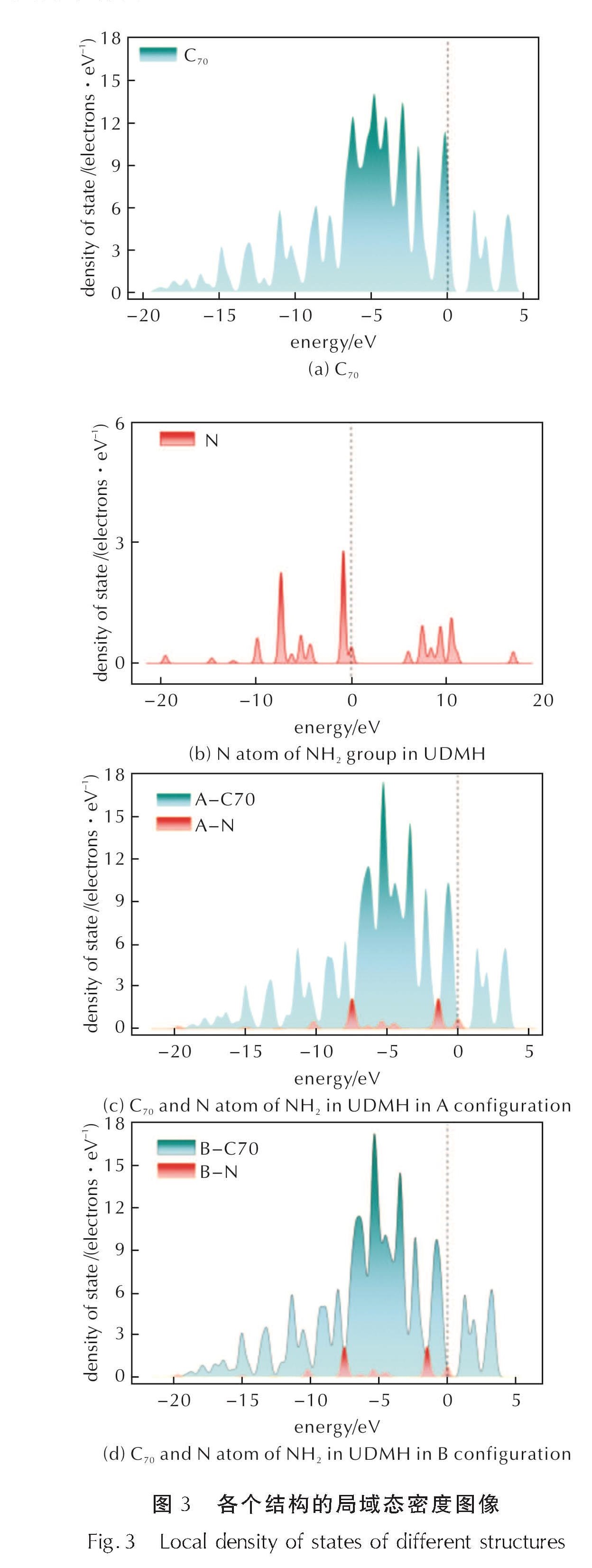
对两种构型中UDMH的吸附能进行计算,结果显示,UDMH在C70上的吸附能分别为:A构型-0.1392eV、B构型-0.1422eV。较低的吸附能表明UDMH分子可能以物理吸附的形式吸附于C70上。通过对纯C70、UDMH以及两种构型进行态密度和分子间距离计算,进一步证明C70和UDMH二者之间的吸附为物理吸附。各个结构的局域态密度(LDOS)如图3所示。由图3(a)、(c)和(d)可知,UDMH与C70形成稳定吸附构型后,C70中的态密度峰发生变化,主要发生在-9~-2eV范围内,说明UDMH和C70之间发生了电子转移。N原子的DOS峰分布于C70分子分布能级的-20~0.5eV范围内,并且在-7~-4eV存在部分重叠。将图3(c)、(d)两种构型的DOS图像相比,在-7~-3eV能级范围内,A构型中C70的DOS峰的峰值比B构型更高,且图2(b)中N原子的DOS峰形和能级并无改变。尽管两种分子间的DOS峰有部分重叠,但是分子间距离较远,不具备形成共价键的条件(采用两个成键原子半径之和的0.6~1.5倍长度来判断共价键的形成与断裂)[27]。
图3 各个结构的局域态密度图像
Fig.3 Local density of states of different structures
2.2 C70与UDMH的相互作用
基于文献研究[9,12]确定无C70作用时UDMH在NO2自由基存在时的初始分解及次级分解路径。将NO2自由基分别放置于CH3和NH2基团并进行抽氢反应过渡态(TS)搜索。计算结果表明,在NH2基团上发生的抽氢反应势垒为34.1kJ/mol,反应热为31.4kJ/mol,在CH3基团上的反应势垒为83.0kJ/mol,反应热为82.9kJ/mol。NH2基团上抽氢反应具有较低的反应势垒,与文献报道一致[11]。
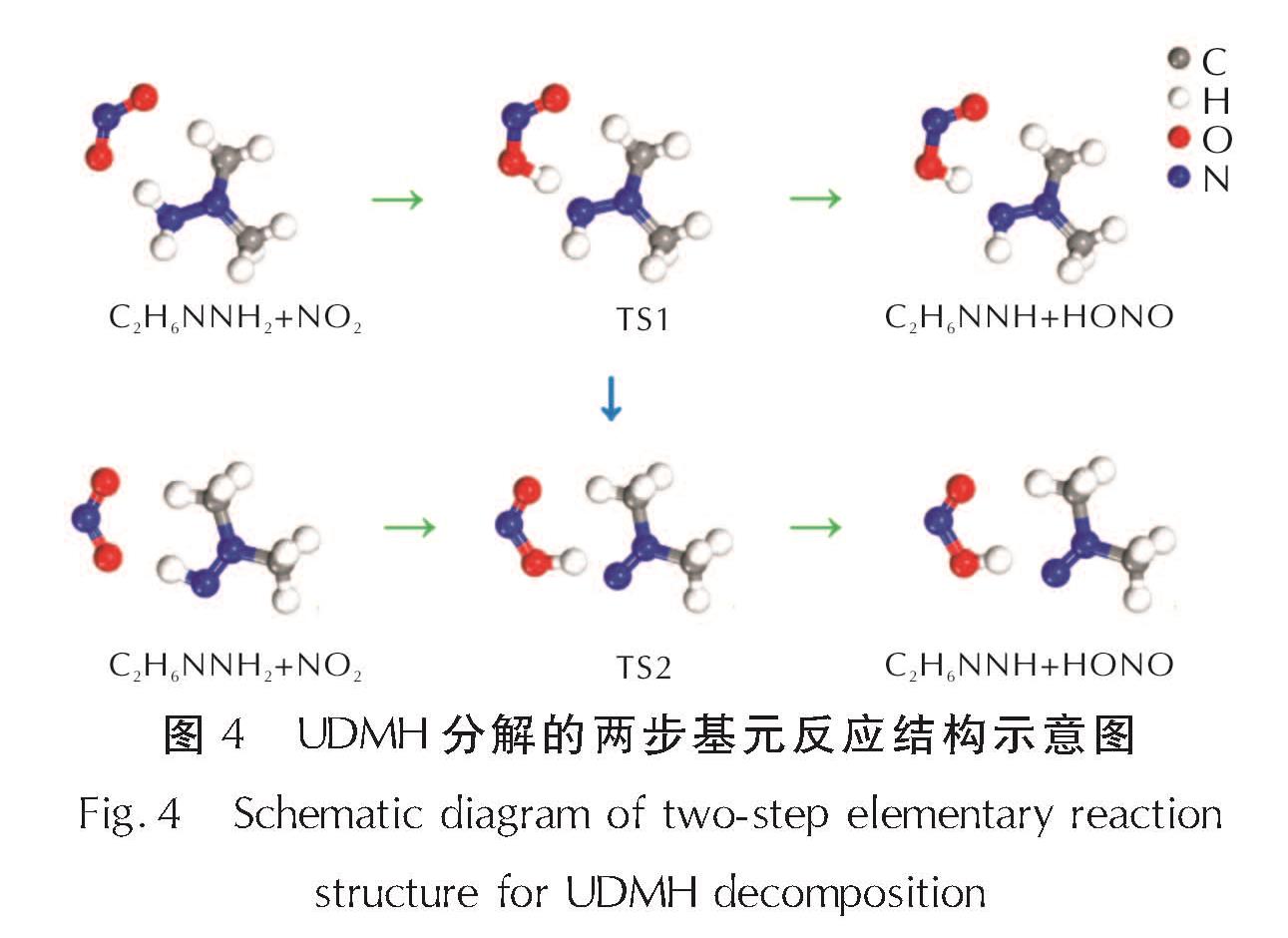
基于上述结果,次级反应仍选择NH基团上发生,以NO2靠近NH基团并发生H原子转移的结构分别作为基元反应的始态(IS)、终态(FS)结构。在NH2上发生的两步抽氢反应中IS、TS、FS结构如图4所示,相应的反应能量曲线如图5所示。UDMH分解初始及次级反应过渡态搜索结果均符合过渡态的搜索结果。
图4 UDMH分解的两步基元反应结构示意图
Fig.4 Schematic diagram of two-step elementary reaction structure for UDMH decomposition
图5 UDMH分解的两步基元反应的能量曲线
Fig.5 Energetic profiles of two-step elementary reaction for UDMH decomposition
当NO2自由基靠近UDMH分子,NH2基团上H原子会发生解离,并转移至NO2自由基上,从而生成C2H6NNH和HONO,反应活化能为34.1kJ/mol,反应热为31.4kJ/mol。第二步抽氢反应以同样的方式进行,经过NO2自由基在NH基团上抽氢,形成C2H6NN和HONO(图4),该基元反应的活化能和反应热分别为28.7kJ/mol和22.8kJ/mol。从数值上分析,第一步抽氢反应的活化能和反应热都明显高于第二步,由此说明第一步抽氢反应对于UDMH分解的发生具有决定性。
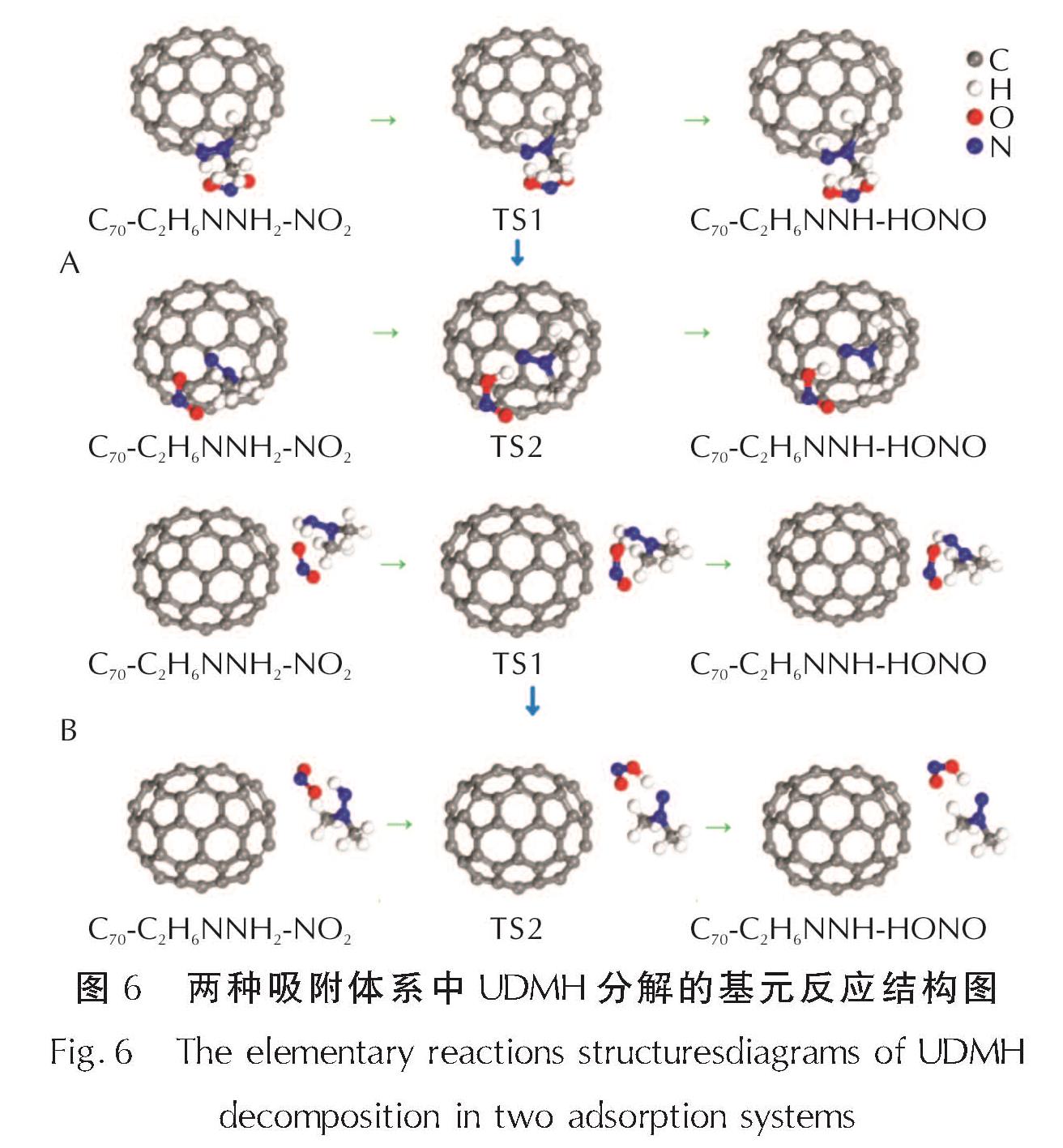
加入C70后,首先研究UDMH分解的起始反应位点可否发生在甲基,故研究了NO2从甲基上发生抽氢的可行性。在尝试优化多种模型之后,都不能得到稳定的IS结构,无法进行后续的过渡态搜索和反应热计算,表明加入C70后,NO2自由基在UDMH甲基上发生抽氢反应的位置仍然在NH2上。进一步计算获得了A、B两种构型与NO2相互作用的稳定构型,如图6所示。
将NH2基团作为UDMH分解初始及次级反应的反应位点,分别选取在此基团附近吸附NO2自由基及对应的H原子转移后的稳定结构作为反应的IS、FS结构。
对NH2基团上两步抽氢反应进行过渡态搜索,得到的反应过渡态均只有一个虚频,证明过渡态的搜索结果正确。C70吸附UDMH体系中两步抽氢反应的IS、TS以及FS结构如图6所示,对应的反应能量曲线如图7所示。
图6 两种吸附体系中UDMH分解的基元反应结构图
Fig.6 The elementary reactions structuresdiagrams of UDMH decomposition in two adsorption systems
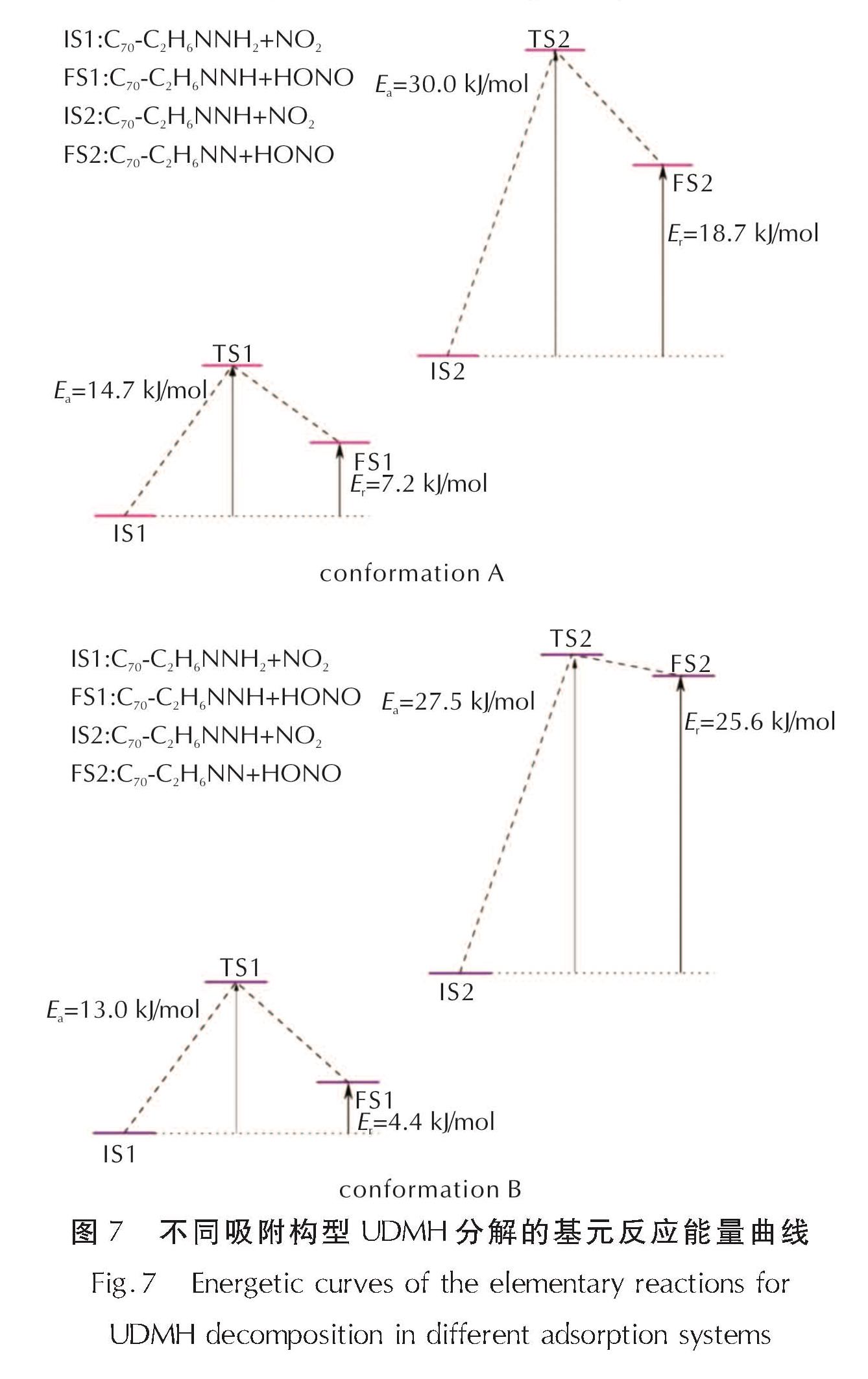
图7 不同吸附构型UDMH分解的基元反应能量曲线
Fig.7 Energetic curves of the elementary reactions for UDMH decomposition in different adsorption systems
当NO2自由基进入反应体系并发生反应时,反应形式与纯UDMH分解相同,均为NO2自由基从NH2基团上抽取氢离子。随着NO2自由基靠近UDMH分子,NH2基团上一个H原子转移至NO2自由基上,形成C2H6NNH分子和HONO。由图7可知,该基元反应在A构型体系中的活化能为14.7kJ/mol,反应热为7.2kJ/mol; 在B构型体系中活化能(13.0kJ/mol)和反应热(4.4kJ/mol)相对更低。在第二步反应发生前,两种构型中的C2H6NNH分子均发生了旋转(图6),但这并没有影响抽氢反应的发生。其中A构型中抽氢反应的反应势垒为30.0kJ/mol,反应热为18.7kJ/mol,而B构型中反应势垒为27.5kJ/mol,反应热为25.6kJ/mol。相较于A构型,第二步反应中B构型反应的活化能更高,而反应热更低。结果表明,在C70存在时UDMH分子分解的前两步基元反应仍为吸热反应,与纯UDMH分解不同的是,C70的加入会使第二步反应的反应势垒和反应热都明显高于第一步。
此外,在对C70吸附体系UDMH第一步分解产物(图9(b)、(e))直接进行优化时,考虑到UDMH中NH2基团在经历了抽氢反应后转变为NH基团,且N—N没有发生改变,故在几何优化前将分子总价态设置为-1进行优化,得到的结果表明该结构可以稳定存在且分子总价态为-1。对第二步反应产物(图9(c)、(f))进行优化时将分子总价态设置为-2、0分别进行优化,结果表明分子总价态设置为0时能量更低,原因可能是在失去了两个H原子后N—N转化为N═N,分子价态重新归零。
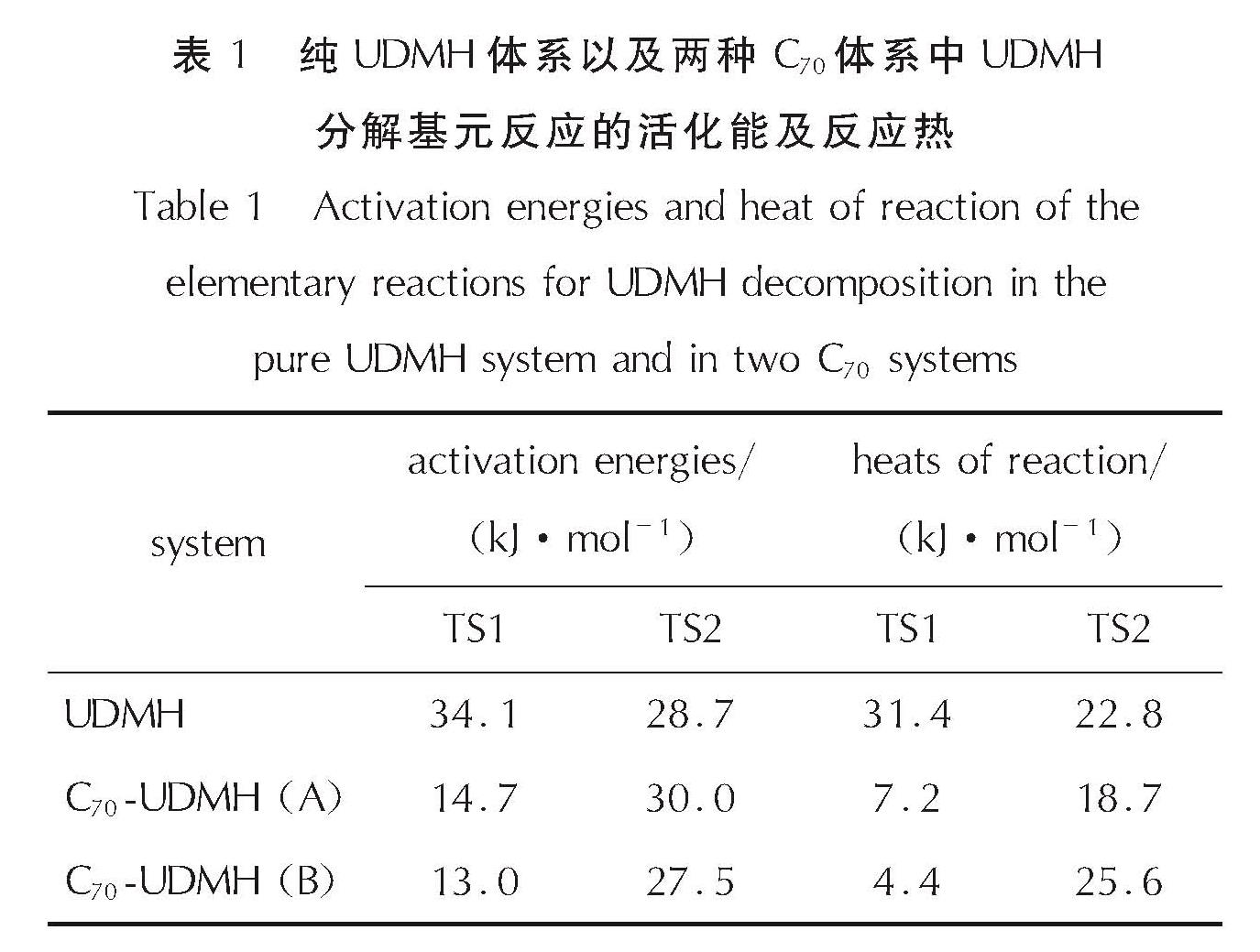
为了从反应热方面具体说明C70用于催化UDMH分解的可行性,表1列出3种体系中两步基元反应的活化能以及反应热数据,相应能量曲线如图8所示。
表1 纯UDMH体系以及两种C70体系中UDMH分解基元反应的活化能及反应热
Table 1 Activation energies and heat of reaction of the elementary reactions for UDMH decomposition in the pure UDMH system and in two C70 systems
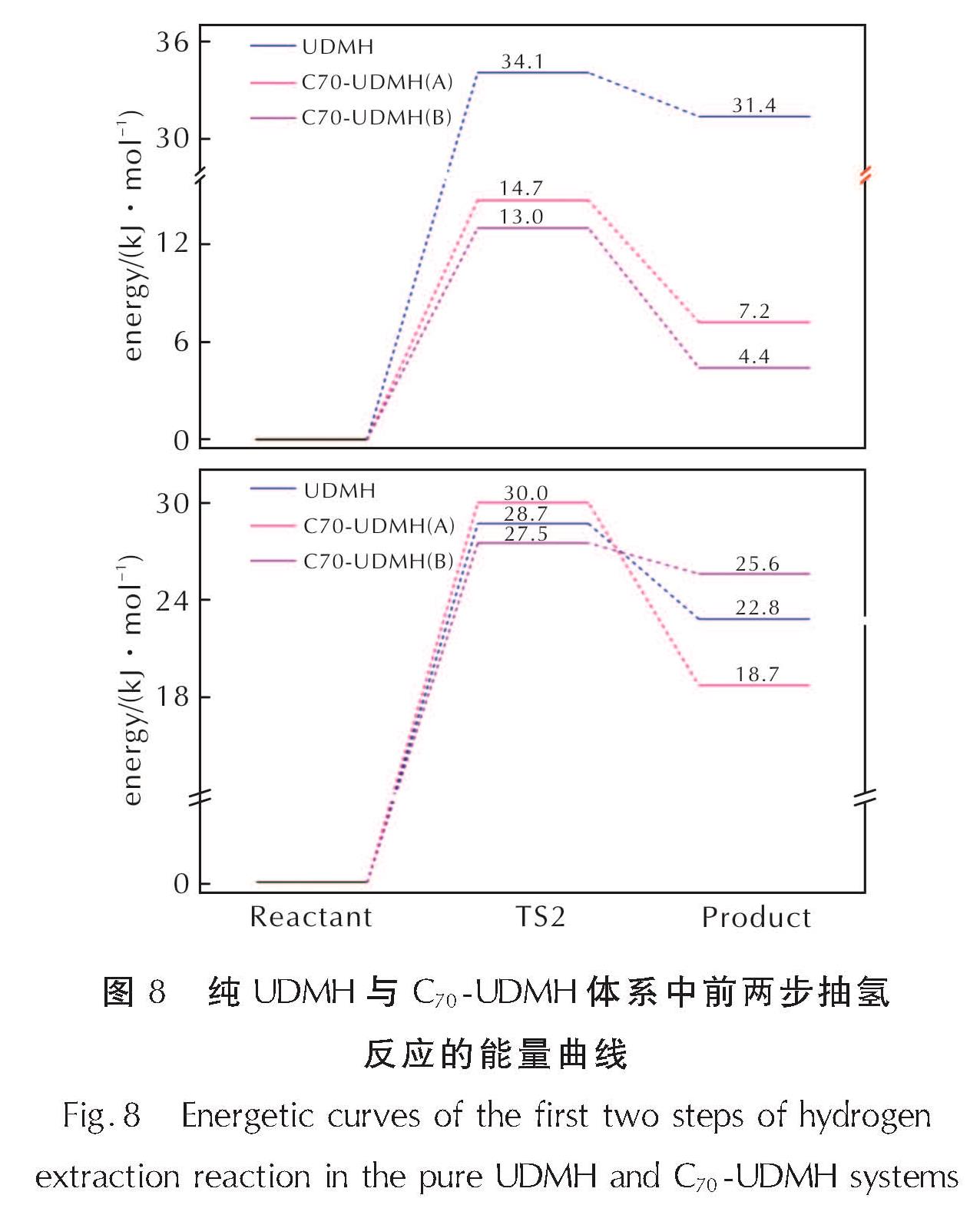
图8 纯UDMH与C70-UDMH体系中前两步抽氢反应的能量曲线
Fig.8 Energetic curves of the first two steps of hydrogen extraction reaction in the pure UDMH and C70-UDMH systemsC70
的加入会显著降低UDMH分解的第一步反应的反应势垒和反应热。相比于纯体系中第一步反应34.1kJ/mol的反应势垒,两种构型中第一步反应的反应势垒分别降至14.7kJ/mol(A)、13.0kJ/mol(B); 反应热从31.4kJ/mol减小到7.2kJ/mol(A)、4.4kJ/mol(B)。这意味着C70的加入对于UDMH第一步分解反应在动力学和热力学上都是有利的。而在第二步反应中,不同的吸附构型C70对UDMH反应的反应热影响不同。计算结果显示,构型A在降低4.1kJ/mol反应热的同时会提高1.3kJ/mol的反应活化能; 而构型B在反应热和反应活化能的改变上与构型A恰恰相反,构型B中第二步基元反应的反应活化能减少了0.8kJ/mol,反应热增加了2.8kJ/mol。结合反应势垒分析,构型A中C70对于第二步反应的发生具有轻微的抑制作用,构型B则有轻微的催化作用。
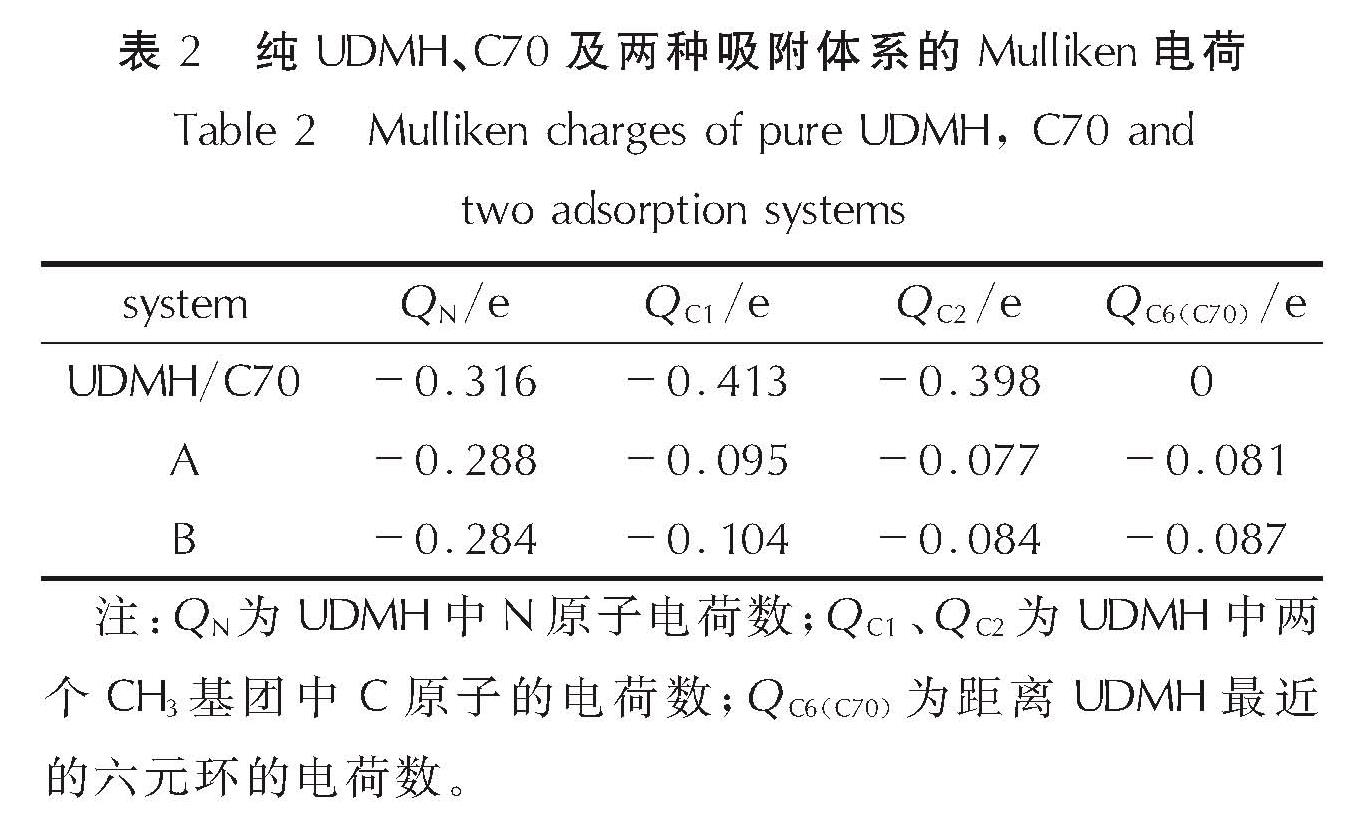
Mulliken布局电荷分布是考察电子结构的一个重要参数,一般来说,电荷值越负,所带电子越多。表2列出了不同构型中UDMH及C70部分原子的Mulliken电荷数。Mulliken电荷分析表明在发生吸附后UDMH中的N、C原子失去部分电子,A构型中N原子失去了0.028e电子,B构型中失去更多的电子(0.032e); 且相较于A构型(0.612e),B构型中的两个C原子失去更多的电子(0.65e)。同时C70中距离UDMH分子最近的一个六元环会得到大部分电子。这说明C70作为催化剂吸附UDMH后,可能会使UDMH分子失去部分电荷,从而使其发生分解反应的能垒降低。
表2 纯UDMH、C70及两种吸附体系的Mulliken电荷
Table 2 Mulliken charges of pure UDMH, C70 and two adsorption systems
综合上述分析,C70的加入对于UDMH的分解具有催化作用。且在第一步抽氢反应中非常明显,A、B两种吸附构型分别降低了19.4kJ/mol和21.1kJ/mol的反应势垒以及24.2kJ/mol、27.0kJ/mol的反应热。计算结果表明,C70的加入会将第一步的反应热和反应势垒降低至比第二步更低的水平。其中,B构型更倾向于降低反应的活化能,A构型对于反应热的降低则更为明显。
当UDMH在吸附体系发生分解时,其中间产物与C70分子之间的距离会不断变化。通过计算获得了两种吸附构型—NH2发生两步抽氢反应的优化吸附构型如图9所示,其中(a)~(c)为A构型的初始状态及中间体;(d)~(f)为B构型对应的结构。UDMH及不同阶段抽氢反应产物与C70团簇分子之间的距离如表3所示。
表3 UDMH以及分解反应中间体中N原子与C70中一个C原子的距离
Table 3 Distances between N atoms in UDMH and its decomposition reaction intermediates and one C atom in C70
相对C70中的碳原子,A构型的UDMH分子在第一步反应结束后轻微向C70分子靠近,N原子和C原子间距离缩短至3.410Å,在下一步反应结束时距离增加至4.266Å。并且UDMH分子会以N—N键为轴发生逆时针旋转。B构型中UDMH与C70在距离上的变化趋势与A构型相同,然而B构型体系中UDMH的分解产物在第一步反应结束相对于C70移动的距离相较于A构型更少,在第二步反应结束相对A构型更多。分子间距离的增加意味着UDMH可能会在其中一步抽氢反应结束后发生脱附,从而进行后续的分解。
图9 (a~f)UDMH及其分解反应中间体; (g)、(h)测距时参考C70选定的C原子
Fig.9 (a—f)UDMH and its intermediates of decomposition reactions; (g)、(h)C atoms selected with reference to C70 during ranging
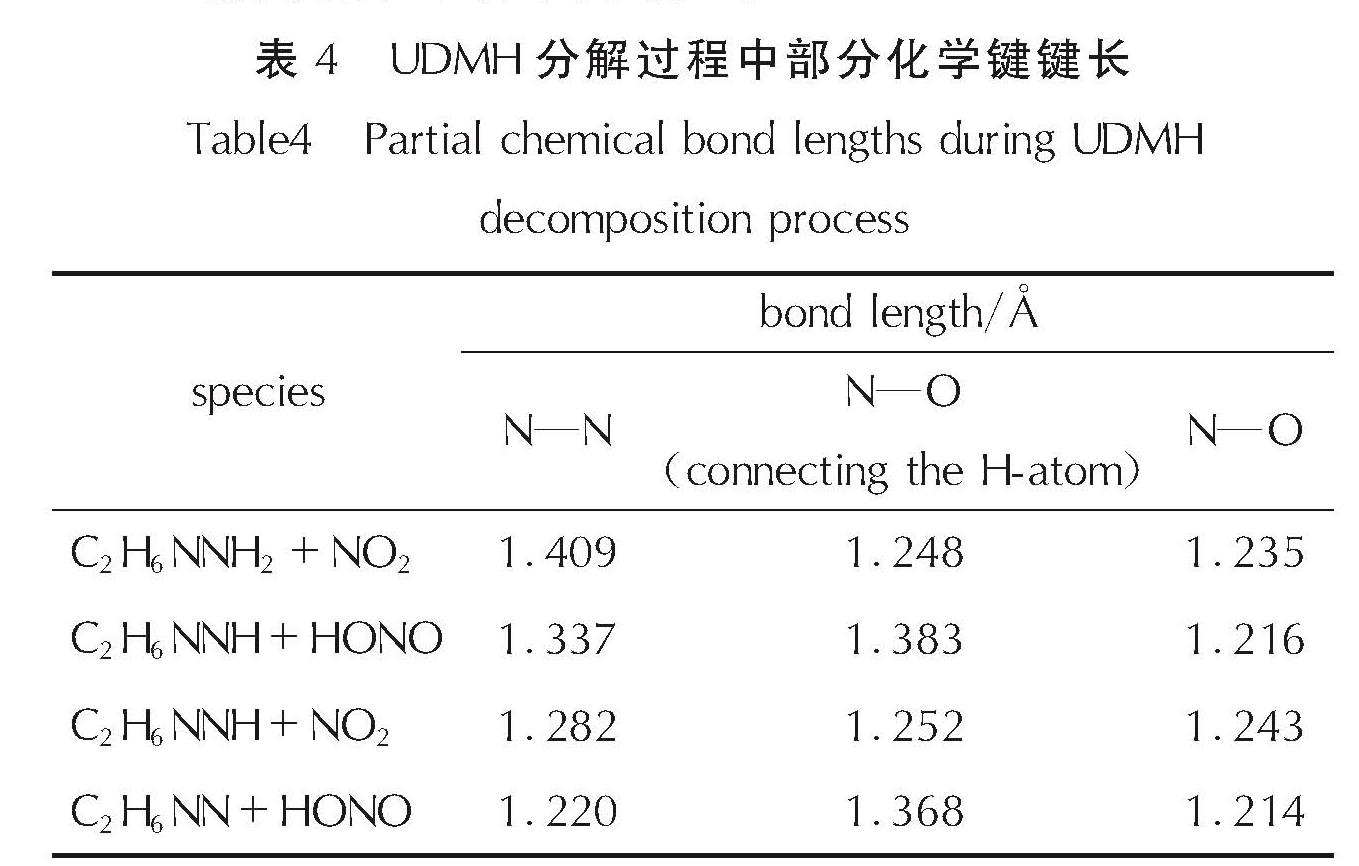
此外,为探究在反应过程中C70对于UDMH分子和NO2自由基及其产物中化学键的影响,测量了不同体系中NO2的两个N═O键以及UDMH中的N—N键的键长,结果见表4。
表4 UDMH分解过程中部分化学键键长
Table 4 Partial chemical bond lengths during UDMH decomposition process
随着UDMH分解反应的进行,N—N键会逐渐缩短,且NO2自由基中不同的N═O键键长也会发生轻微的变化。在NO2自由基进入体系时,其中两个N═O键键长在1.235~1.252Å; 而形成HONO自由基后,连接H原子的N═O键被拉长至1.368~1.383Å,没有连接H原子的N═O键会缩短至1.214~1.216Å。并且不同体系中化学键的键长在数值以及变化趋势上几乎相同。这说明在UDMH分解过程中,C70对于UDMH分子及NO2、HONO自由基中化学键的影响并不明显。
进一步研究发现,纯UDMH经历两步抽氢反应并向体系中加入NO2后,(CH3)2NN中的N原子会吸收自由基中的O原子,转化为(CH3)2NNO,随着NO2含量的增加,N—N键断裂生成(CH3)2N。而在C70体系中却没有发生这种现象,此时UDMH分解产物与C70的分子间距离达到4.27Å(A)、4.86Å(B),因此推测可能发生了UDMH脱附,C70对于后续反应的影响需要进一步深入研究。