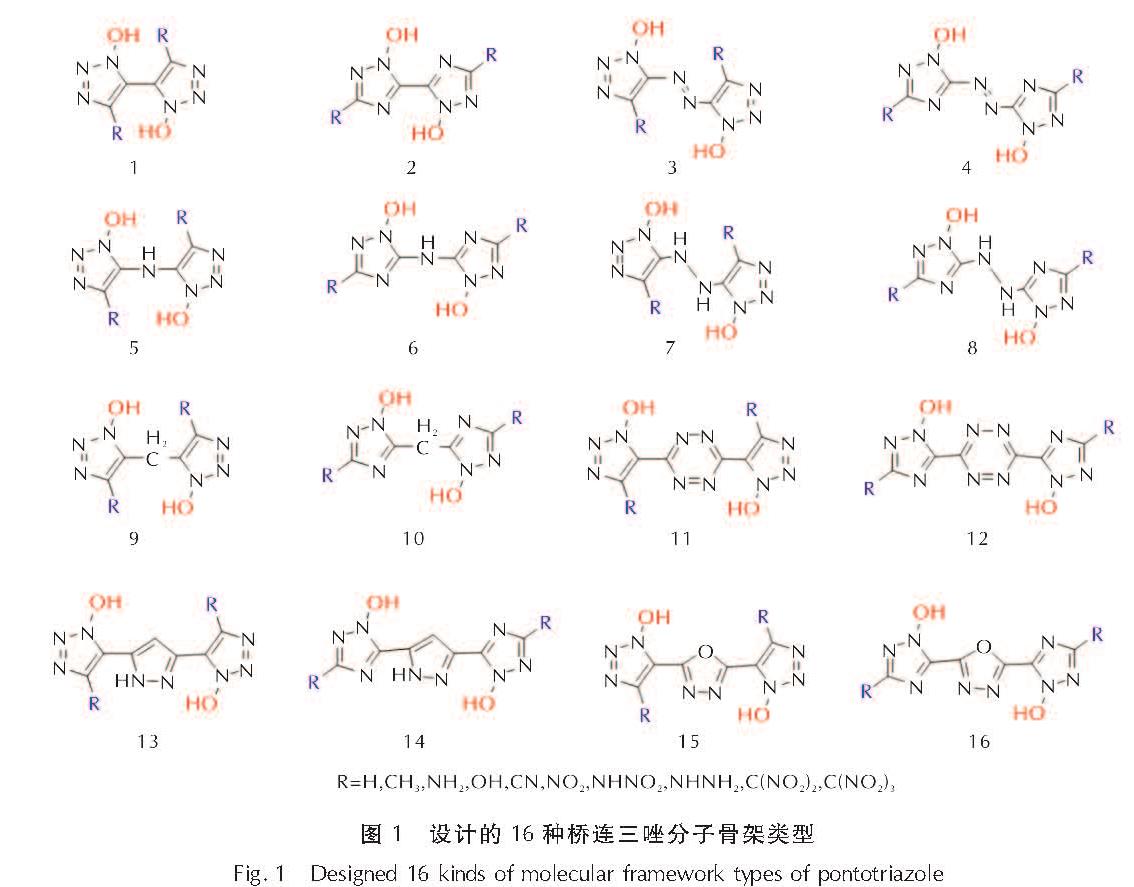
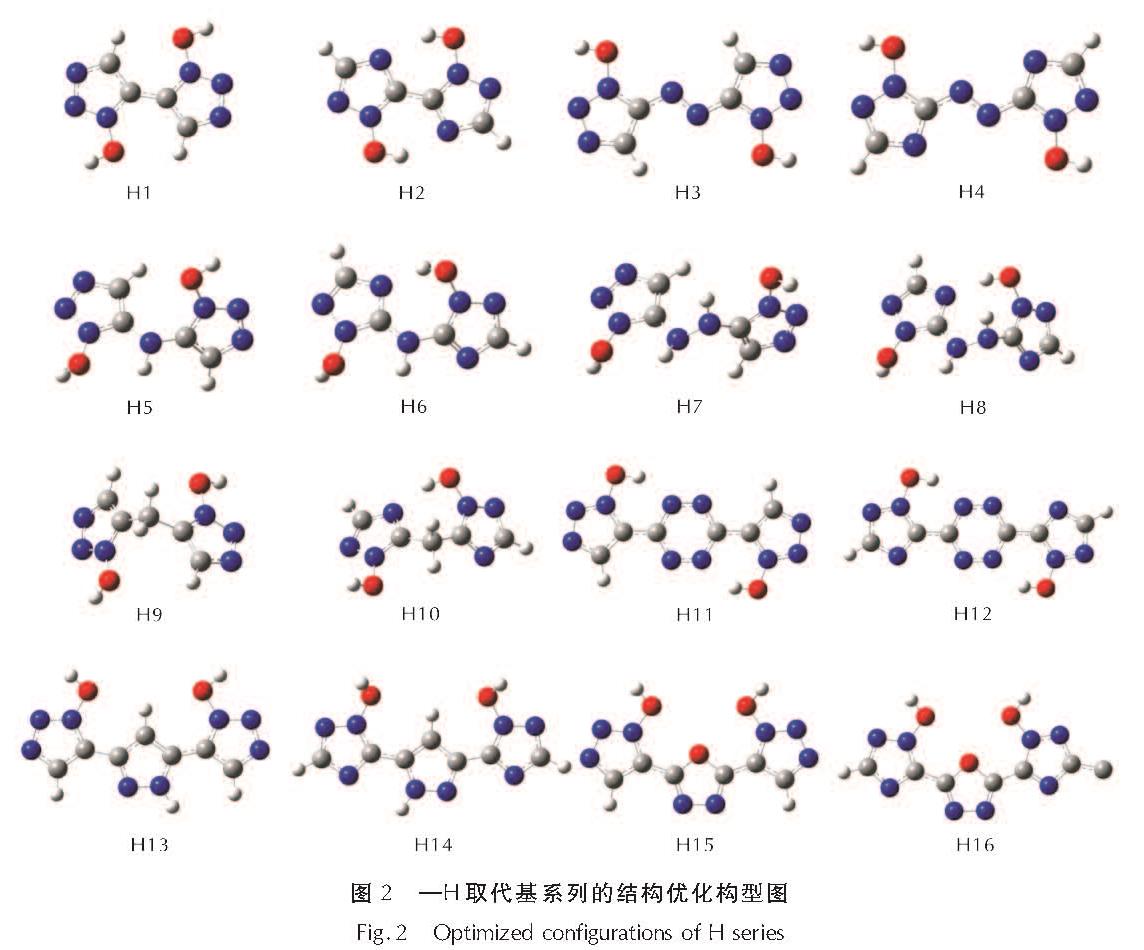
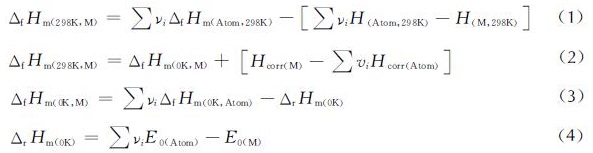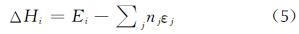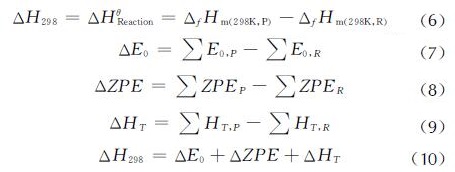2.1 几何优化
为了选取合适的计算模型,本研究中160个分子的初始结构都设置为能量更低的反式构象(反式1,2,3-三唑、反式1,2,4-三唑)。几何优化收敛后,桥基为H(1)、—N═N—(2)、四嗪(6)、吡唑(7)、呋咱(8)的化合物,环骨架仍保持和初始结构相似的平面构型,而其他桥连化合物(桥基为—NH—、—NHNH—、—CH2—),在桥基处有“折角”,使得两个唑环无法共平面。
由吡唑(7)、呋咱(8)桥接的双环三唑化合物,均翻转为顺式构型,说明由这两种桥基连接的化合物更趋于顺式构象; 而由H(1)、—N═N—(2)、—NH—(3)、—NHNH—(4)、—CH2—(5)、四嗪(6)连接的双环三唑化合物趋于反式构象。本研究的160个分子的几何优化结构图见支撑材料中图S1~S9,图2为—H取代基系列的几何优化构型图。
图2 —H取代基系列的结构优化构型图
Fig.2 Optimized configurations of H series
以H(1)、—N═N—(2)、四嗪(6)、吡唑(7)、呋咱(8)为桥基的桥连双环三唑环,环骨架仍保持和初始结构相似的平面构型,表明当唑环与桥基共轭共平面时,分子的稳定性更高; 此外,—N═N—、四嗪、吡唑、呋咱这类桥基,由于体系中引入了N—C、N—N键,将有利于提高含能化合物的爆炸性质[23]。而—NH—(3)、—NHNH—(4)、—CH2—(5)桥连化合物中,两个唑环面存在“折角”,两个唑环和桥基之间由于电子排斥作用而呈异面构型。这可能是因为—NH—与—NHNH—的孤对电子在sp3杂化轨道,无法与唑环的游离电子共轭,而—CH2—没有多余的孤对电子或空轨道; 这三种桥基的H原子处在sp3杂化轨道,受空间排斥的影响,两个唑环与桥基均呈异面构型。
由吡唑(7)、呋咱(8)桥接的双环三唑化合物均翻转为了顺式构型,说明由这两种桥基连接的化合物更趋于顺式构象; 这可能因为反式构型下,吡唑上的H与唑环羟基上的O的距离为1.55605Å,相比于顺式构型下吡唑H与羟基O的距离为2.47471Å,原子间空间斥力更弱; 呋咱系列化合物在顺式构型下取代基对称向两边伸展,而反式构型下取代基受呋咱的空间排斥。
2.2 不同方法计算的生成热对比
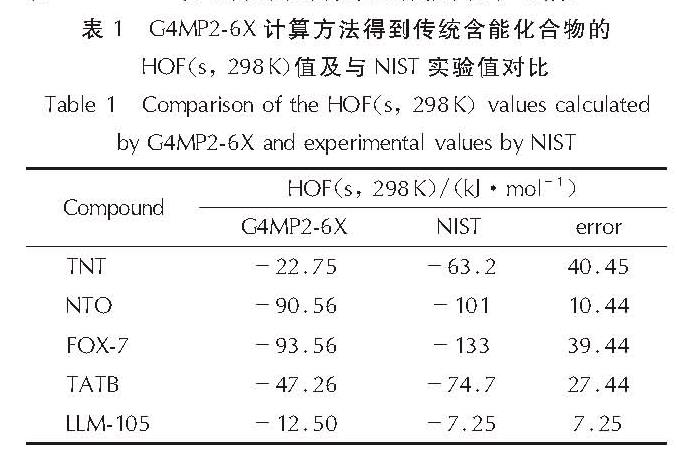
用G4MP2-6X方法计算了常用炸药TNT、NTO、FOX-7、TATB与LLM-105的固态生成热,结果见表1。与NIST网站查阅的实验值对比,误差不超过40kJ /mol,证明G4MP2-6X方法预测的标准生成热结果可信。
表1 G4MP2-6X计算方法得到传统含能化合物的HOF(s, 298K)值及与NIST实验值对比
Table 1 Comparison of the HOF(s, 298K)values calculated by G4MP2-6X and experimental values by NIST
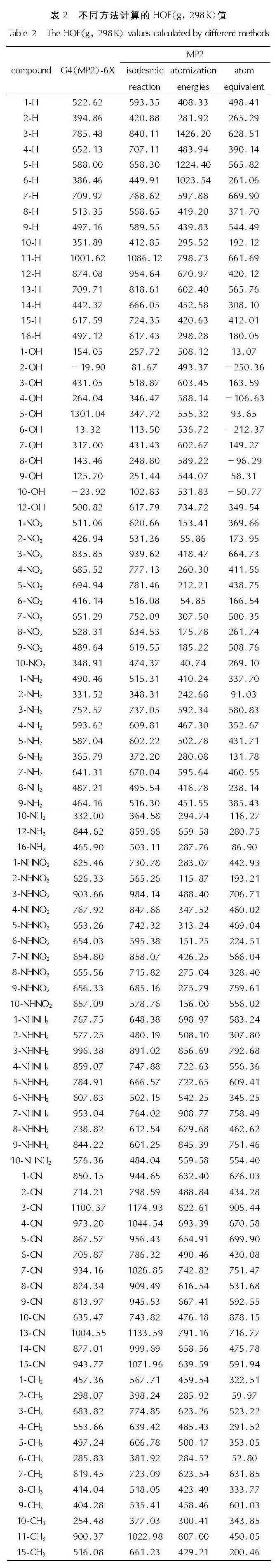
在B3LYP/6-311G(d,p)//MP2/6-311++G(d,p)理论水平下,分别用原子化能法、原子当量法与等键反应法对160种设计的含能分子的标准摩尔生成热进行计算,并与G4MP2-6X方法直接读取的标准摩尔生成热值进行对比,结果如表2所示。
表2 不同方法计算的HOF(g, 298K)值
Table 2 The HOF(g, 298K)values calculated by different methods
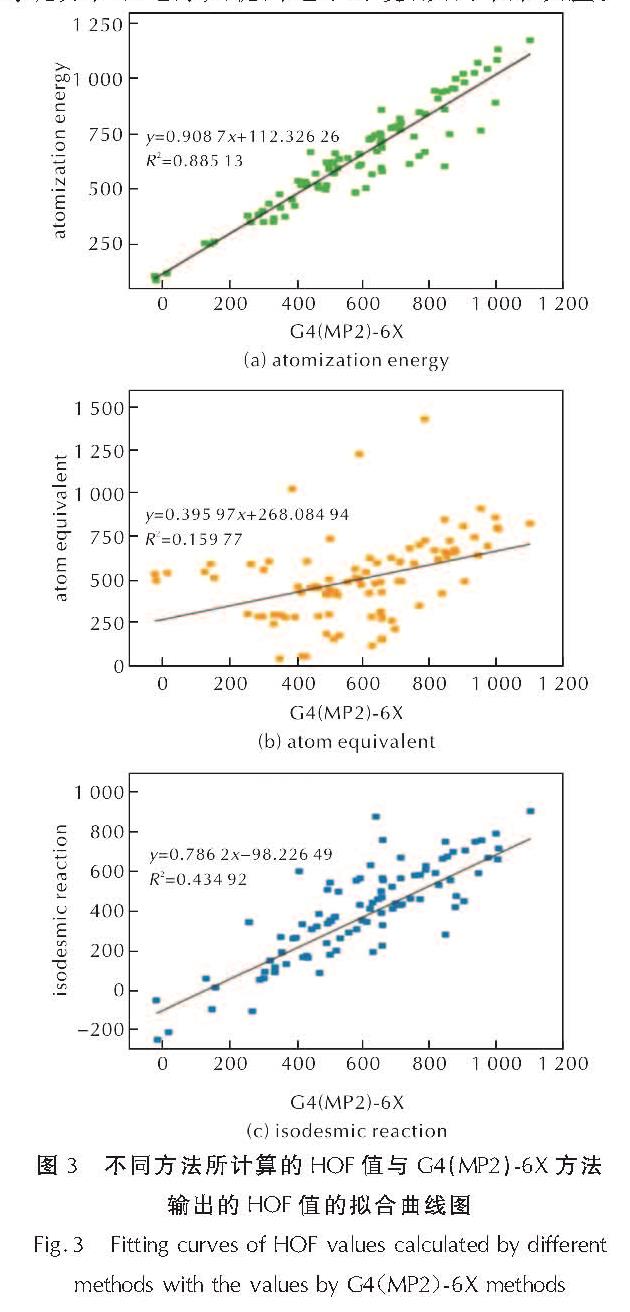
为了对比3种辅助反应(原子化能方法、等键反应方法与原子当量方法)求得的标准摩尔生成热与G4(MP2)-6X方法结合原子化能方法输出的标准摩尔生成热的相关性,分别绘制了3种方法与G4(MP2)-6X方法的线性相关图,并拟合了辅助方法与G4(MP2)-6X方法的生成热的一元回归方程,如图3所示。其中,原子化能方法的相关性最高,相关系数为0.88513; 等键反应方法次之,相关系数为0.43492; 原子当量方法的相关性最低,仅有0.15977。对于本研究的160个含能分子,等键反应的相关性较低,可能是因为以—H(1)、—N═N—(2)、四嗪(6)、吡唑(7)、呋咱(8)为连接基团的分子,它们的共轭环骨架在等键拆分时被破坏,并且无法利用已有气相实验生成热数据的小分子建立等键方程以还原共轭的电子环境,从而带来误差。
图3 不同方法所计算的HOF值与G4(MP2)-6X方法输出的HOF值的拟合曲线图
Fig.3 Fitting curves of HOF values calculated by different methods with the values by G4(MP2)-6X methods
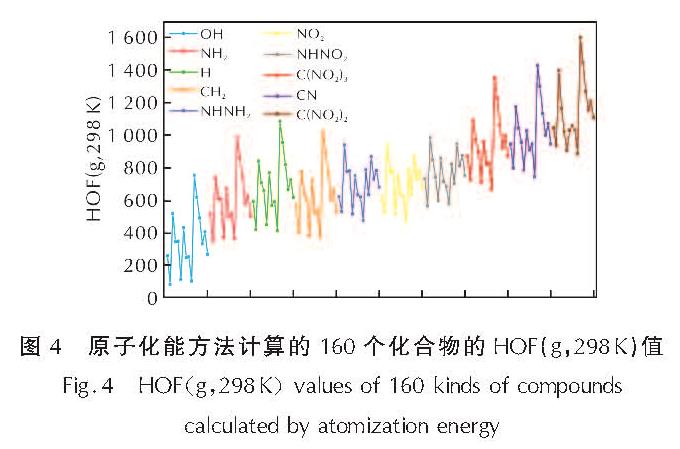
图4为按照基团分类的160个化合物的HOF值(原子化能方法计算)。
图4 原子化能方法计算的160个化合物的HOF(g,298K)值
Fig.4 HOF(g,298K)values of 160 kinds of compounds calculated by atomization energy
由图4可知,取代基—C(NO2)2、—C(NO2)3、—NHNO2和—CN显著提高了分子的生成热,而—OH与—NH2取代基系列的生成热低于—H系列。—C(NO2)2的氮含量与CN键数量都不如—C(NO2)3,但HOF值更高,这可能因为偕二硝基基团呈平面构型,共轭的结构使得生成热提高。同一桥基连接的联三唑同分异构体(联1,2,3-三唑,联1,2,4-三唑),桥连双1,2,3-三唑的HOF更大,这可能因为1,2,3-三唑的生成焓(272kJ mol-1)比1,2,4-三唑(109kJ mol-1)高。对于10种不同取代基,四嗪联三唑分子骨架都具有最高的HOF值,可能因为四嗪与三唑环共轭,分子生成热提高。