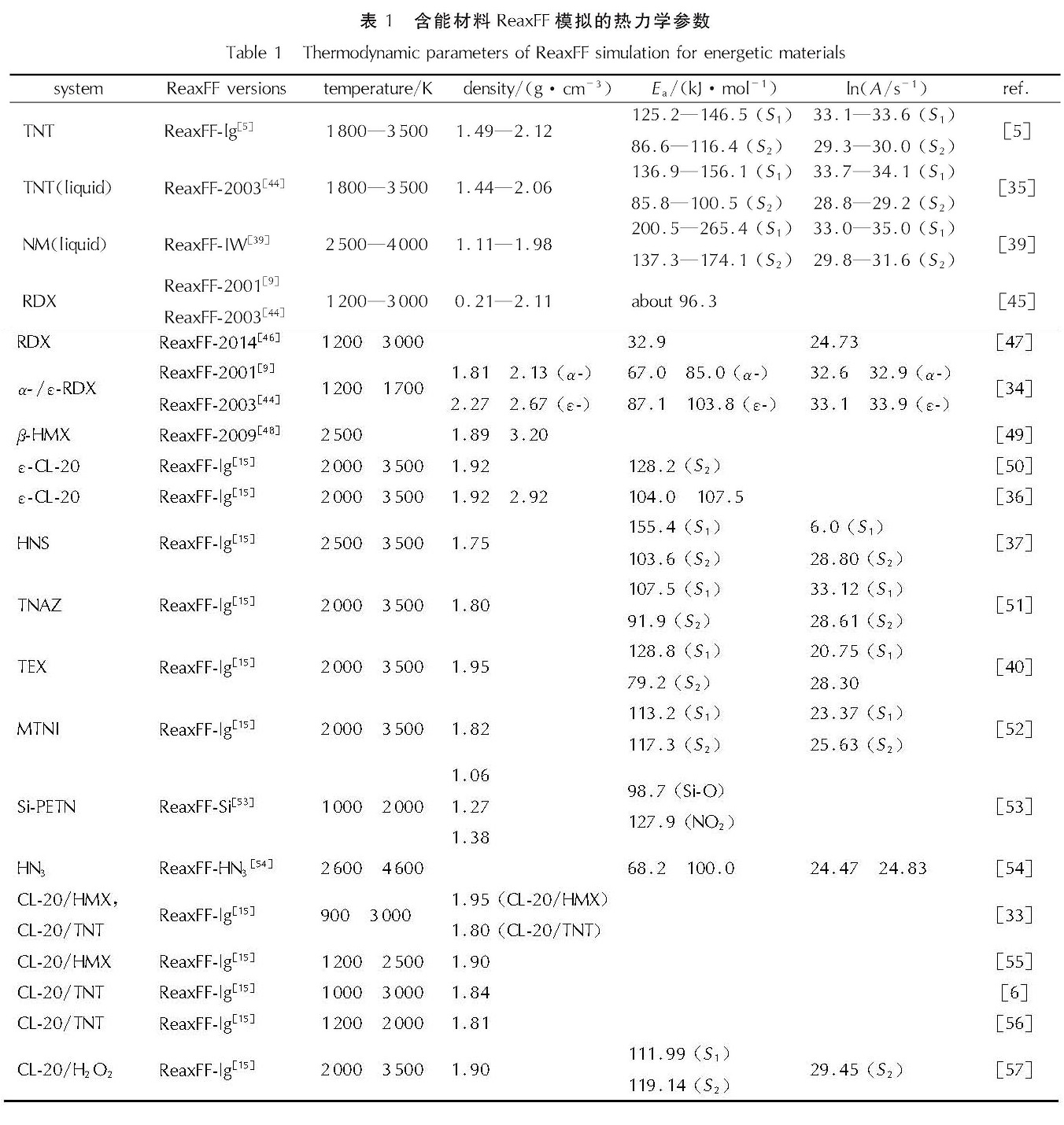
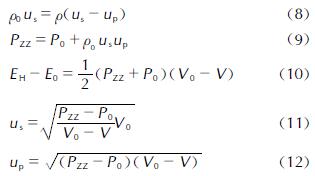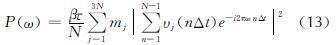1.1 ReaxFF反应力场介绍
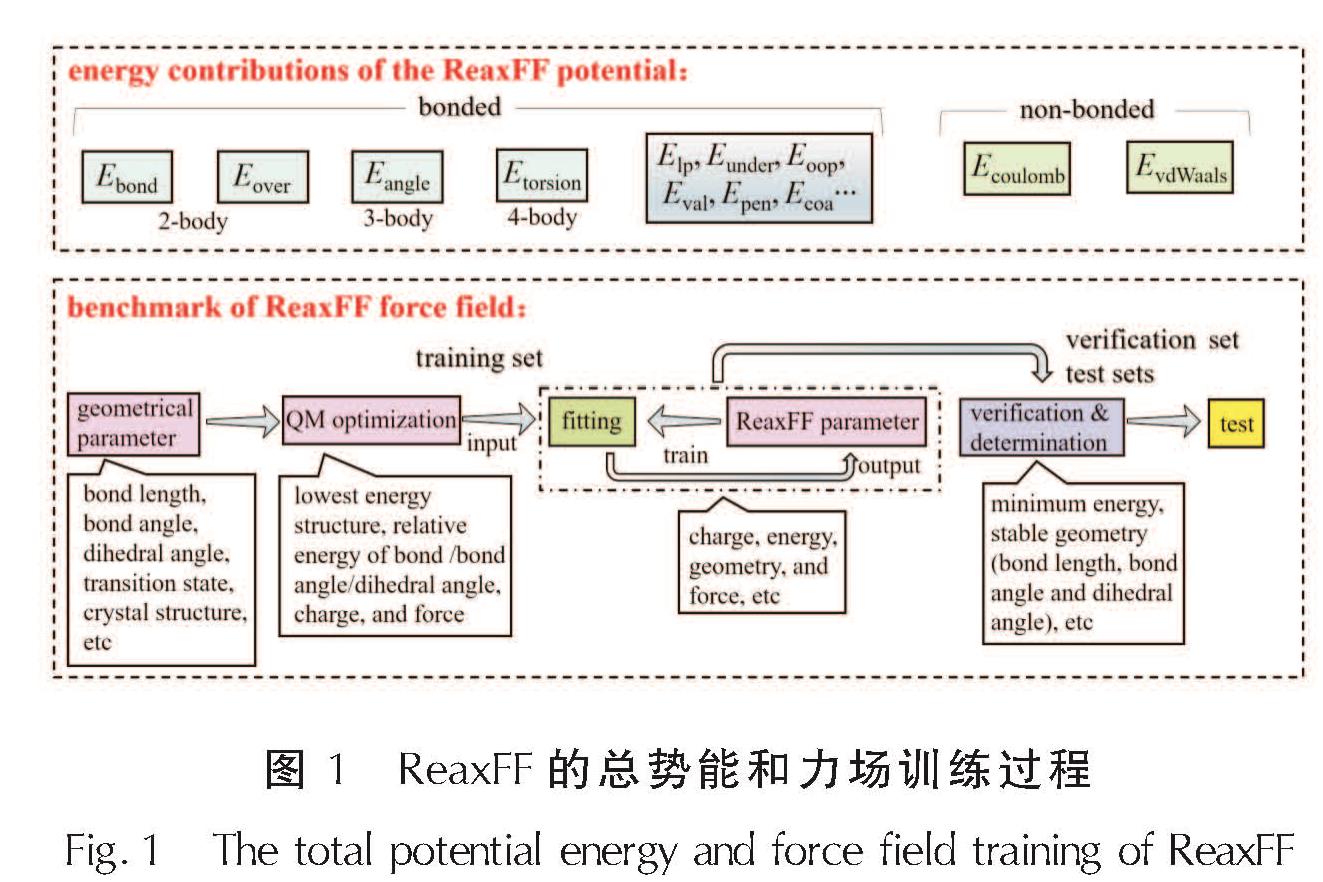
ReaxFF采用结合可极化电荷描述的键级形式来描述原子之间的反应性和非反应性相互作用,键级为使用经验公式直接从原子间距离计算得出。ReaxFF各能量项的总结和其力场训练过程如图1所示。此外,为了弥补ReaxFF力场在有机晶体中对长程vdW相互作用低估的缺点,Liu等[15]引入基于低梯度(Low-Gradient,lg)模型的修正项对色散作用进行修正。
图1 ReaxFF的总势能和力场训练过程
Fig.1 The total potential energy and force field training of ReaxFF
ReaxFF反应力场虽具有强大的化学动力学仿真能力,但毕竟是量子化学方法的一种简化和近似。为了最大程度地精确再现量子力学相互作用下的化学反应过程,ReaxFF反应力场以经典相互作用对其近似只能是特化的,因而具有很差的迁移性。对于不同类型的体系,即使其中某些体系具有相同的物质,也不得不开发出多套力场参数分别使用。例如,有机炸药和生物体系均有大量有机官能团和水、二氧化碳等基础物质,却不能共用一套ReaxFF反应力场参数,因为两种体系分别需要对热分解反应和肽水解反应进行特化,以使精度达到最低要求。而对于研究者而言,一旦获得了目标体系的一套优良的ReaxFF反应力场参数,则代表着其化学动力学研究能力几乎立即从单分子层面达到了介观层次,足以针对界面、孔隙、缺陷、边界、晶格等微观结构效应进行还原,大量的实验现象得以重现,新的机制得以挖掘。
由于对各种特定体系开发ReaxFF反应力场参数的巨大需求,力场参数训练一直都是研究的热点领域。与经典非反应力场不同,ReaxFF反应力场包含了大量的两体、多体相互作用势,力场参数集庞大,训练集包含的化学响应种类多。换言之,训练一套ReaxFF反应力场参数,是一个N维(N=103数量级)M目标(M=102数量级)的最优化问题,因而其训练具有很大的难度。数十年来,一直有新的、更有效的ReaxFF参数优化方法被开发出来,称之为“力场开发的开发(Development of the development of forcefield)”。其发展如图2所示,最初创立的方法为序贯抛物线参数插值法(sequential parabolic parameter interpolation method,SOPPI)[16]。
图2 ReaxFF反应力场优化方法的发展
Fig.2 Development of ReaxFF reaction force field Parameter optimization method
SOPPI对训练集参数需逐一执行单参数搜索,直到满足指定收敛标准为止。SOPPI方法虽简单,但有以下几个缺陷:(1)容易陷入局部最小值;(2)参数量增多时,会使单参数优化量急剧增加;(3)收敛成功与否和初始参数猜想以及各参数优化顺序密切相关。针对SOPPI的缺陷,全局优化方法被使用并展示出高效、自动化的优势,如遗传算法(GA)[17-18]、模拟退火法(SA)[19-20]、进化算法(EA)[21]、粒子群优化(PSO)[22]和机器学习(ML)[23]。全局优化可作为初步优化,在优化接近收敛时,可结合简单的SOPPI方法。
1.2 多目标进化策略
目前,大多数ReaxFF参数是通过执行单一罚函数(penalty function)优化的,罚函数又是通过在指定参数的训练集中评估不同量获得误差的加权组成。在权重的分配上难免需要人为干预来调整优化过程,尤其是涉及到不同维度下组合量的权重,例如,键长、电荷和构型能等。Larentzos和Rice等[24-26]首先引入基于帕累托最优(Pareto dominance)的多目标方法,无需对ReaxFF各参量指定任何权重。在运算中,该方法产生一系列解决方案,原则上帕累托边界可以通过使用随机变化的权重进行重构并使其不断进化,对应于各种任意权重的所有可能值。
多目标进化策略(MOES)框架是一种无需指定目标的后验方法,结合了进化策略(ES)和数据包络分析方法(data envelopment analysis, DEA),被用于训练ReaxFF力场参数[27]。在MOES中,基于帕累托最优提供了一种用于平衡各种不相称量(键长、键角和二面角等)的自动运算策略,从而消除了人为设定权重对优化的影响。力场训练过程包括:(1)准备训练集和初始ReaxFF参数。其中,训练集包含足够多的从头算数据和实验值,如电荷、几何构型(键长、键角和二面角)、生成热、振动频率和能量等,从文献中获取现有力场参数作为初始ReaxFF参数;(2)将初始参数优化的能量和结构特性与训练集进行比对并产生误差,随后输入到MOES中;(3)MOES对误差进行评判并重新创建新一代的力场参数,有相称维度的类似参考数据点被分组在一起,目标函数(误差)的计算公式如下:
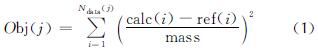
式中:j表示目标。
反复执行上述过程直至达到收敛标准。获得的各项ReaxFF收敛参数需经基于帕累托分析的动力学处理得到最优参数。
运用MOES方法对RDX、FOX-7、HMX、TATB、CL-20和PETN的力场参数进行拟合,并获得的最优晶胞参数。优化后的力场可较好地描述这几种炸药的晶格参数、晶体内分子取向和分子结构。这些炸药晶格参数最大误差的绝对值依次为2.28%、3.89%、1.60%、10.30%、5.15%和3.90%。由于TATB中存在强氢键,在长程相互作用下晶格参数的偏移误差导致TATB的密度误差增大,然而ReaxFF-lg模型中的分子更平坦,许多氢键与分子平面呈90°夹角。综上,MOES方法的优势在于:相较于传统的单一目标参数搜索,依托帕累托最优方法分析产生无需人为指定权重的多个有效力场方案,通过对这些有效力场的评估得到最优力场。因此,MOES方法适合用于力场的开发。Larentzos和Rice[28]还利用这种可迁移的拟合力场策略将ReaxFF-lg力场扩展到硝基甲烷的研究。
1.3 增强的粒子群优化算法
粒子群优化(particle swarm optimization, PSO)算法也被用于训练ReaxFF力场,是一种用于处理随机群体的演化算法,可避免陷入最小化可用于优化非线性、不可微函数的全局搜索方法。PSO的优化工作开始于多组猜测(或随机生成)的力场参数,将每组待优化参数视为解空间中的一个点粒子,而每次迭代产生的参数变化量数组则被视为该粒子的移动速度,功能主要体现为以下3点:(1)同时接受优等和劣等解决方案,在提高随机搜索效率的同时也避免过早收敛的问题;(2)允许同时存在大量潜在的解决方案,从而提高了在盲搜过程中找到目标的概率;(3)与模拟退火不同,PSO在各搜索个体之间共享信息。搜索个体的位置取决于先前行为的惯性项和个体自身的经验参考。个体的速度和位置表示为:
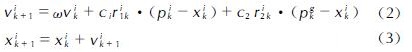
式中:vik和xik分别为个体i在迭代k时在解空间中的位置和移动速度; pik为个体i的最佳位置; pgk为群体的全局最佳位置; 系数c1和c2为个体和全局项的恒定权重; ω为惯性因子,被用于抑制个体的动量,以防止在优化过程中速度无限增长; 参数ri1k和ri2k表示在[0,1]范围内的均匀分布采样的独立随机标量。
在大多数情况下,可分离函数在旋转下变得不可分离。使用随机标量(而不是向量)使得算法对搜索空间的旋转保持不变(rotation-invariant PSO,RiPSO)[29]。该算法将在可分离和不可分离函数中提供一致的性能。然而,在处理复杂的分子力场时RiPSO算法明显性能不佳[30]。这是由于RiPSO算法缺乏多样性,进而导致群体各成员轨迹塌陷到低维子空间中,即线搜索。Furman等[22]对标准RiPSO算法进行直接的扩展,各向同性的高斯变异(GM)算子被用于增强其全局搜索能力,防止其退化为线搜索,因而被命名为具有高斯突变的旋转不变粒子群优化(RiPSOGM)。变异算子的作用是用群体中最佳个体的变异版本替换当前个体,见下式:
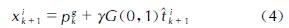
式中:个体的新位置xik+1被设置为以最佳粒子位置pgk为中心的各向同性随机值。步长是从具有零均值和统一的标准偏差的高斯分布G(0,1)中采样的。尺度参数γ与域边界相关,并控制分布的宽度。单位向量t^kk+1通过在d维超球面的表面上均匀拾取点来确保位移的分布是各向同性的。
使用RiPSOGM对色散作用力场参数进行优化,通过DFT-TS计算了HMX、TATB、PETN、FOX-7和NM的状态方程。参数训练中的目标函数是以加权误差平方和的形式表示:
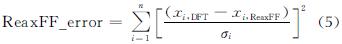
式中:xi,DFT为参考值; xi,ReaxFF为计算值; σi为为训练集中的每个优化目标指定的权重。
Furman等[15]在使用RiPSOGM优化后获得的ReaxFF-lg预测的升华热,并与Liu等拟合的ReaxFF-lg力场作对比。通过对PETN、NM和TATB的测试,发现使用RiPSOGM优化的ReaxFF-lg力场总体上达到了良好效果。对于反应热和活化能等关键反应性质,以TNT、RDX、HMX和CL-20四种典型炸药为例,ReaxFF虽然不能给出准确值,但能获得正确的定性排序,如图3所示。
图3 实验和ReaxFF得到的典型炸药反应热(Er)和活化能(Ea)
Fig.3 Reaction heat(Er)and activation energy(Ea)of typical explosives obtained by experimental and ReaxFF methods
1.4 基于神经网络的反应力场
基于神经网络的反应力场是依托了大量的ReaxFF-MD模拟数据,用以拟合原子间势能的方法[31]。这些势能模型与基础物理量无关,将局部原子环境与能量/力建立关系。Yoo等[32]提出了一种数据驱动的力场:神经网络反应力场(NNRF),并较好的描述硝胺类炸药(eg. RDX、HMX、NM)的复杂化学反应。训练集是由密度泛函中的Perdew-Burke-Ernzerhof和处理London色散作用的D2校正计算得到的大量电子结构。双向不同的反馈回路(凝聚相和单分子系统)使得训练集更加丰富。NNRF方法使用二体和三体对称函数与结合局部原子环境的映射。此外,还通过预测测试集中的解离曲线、能量和作用力来验证。
NNRF的输入层是以原子为中心的加权高斯对称函数,公式(6)和(7)中分别为从分子几何结构中提取的径向(wGradi)和角(wGangi)两种高斯对称函数。

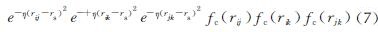
式中:Zj是用于区分不同元素的原子量; rs、η、λ和ξ是要指定的超参数; fc(r)是截止函数,其在5Å的截断半径之外平滑衰减为零。
独立的神经网络被用来描述每个元素的能量和力。每个神经网络由一个输入层(42个节点)、两个隐藏层(各50个节点)和一个输出节点(原子能量)组成。其中,将数据随机分为用于优化神经网络的训练集(包含90%的数据)和用于评估收敛性并避免训练期间过度拟合的验证集(10%)。在较宽温度范围内(1500~2500K),NNRF方法被应用到RDX的高温分解的复杂化学性质中,并得到与实验相近的分解产物和活化能。RDX分解会产生大量中间体和产物分子,如CH2O、HCO2、HON、HONO、HOCN、HCN、CO、CO2、NO2和N2等。通过对比PBE-D2、ReaxFF-2014、ReaxFF-2018和ReaxFF-lg以及实验参数计算的形成能[32],发现NNRF的形成能较好地吻合实验值。
NNRF利用神经网络捕获具有径向和角度描述符的短程相互作用(<5Å),并采用ReaxFFs的van der Waals和Coulomb相互作用参数化表达远程相互作用(<10Å)。使用NNRF的迭代训练研究复杂化学具有DFT级别的准确性,并可转移至其他硝胺炸药(HMX、NM、TNT、TATB、CL-20和PETN),为含能材料的大规模热分解和冲击动力学提供了新的解决方案。然而,由于NNRF力场针对体系的几何结构给出力和能量数值的过程是“黑盒”式的,缺乏明确的相互作用形式表达式,难以明确判定化学反应或进行统计,且体系的某些热力学参数还无法从模拟中获得(如熵和自由能),有待进一步解决和发展。